
生物大数据可视化的现状及挑战
分类:计算机职称论文
时间:2022-03-06
摘要 在过去的10年中, 以基因组学、医学遗传学和神经信息学等为代表的生命科学各研究领域, 以前所未有的增长趋势, 积累了海量的数据信息. 这些数据类型复杂、数量庞大, 其中蕴含的价值更是不可估量. 通过传统的处理手段, 难以理清海量原始数据中错综复杂的关联信息. 而针对生物大数据的可视化研究, 将有利于科研人员对复杂数据进行多角度观察并获取有效信息. 生物数据量越大, 复杂性越高, 可视化在生物有效信息挖掘方面发挥的作用就越大. 本文通过例举若干生物机构中心现存的数据规模和数据增长速率, 说明生物研究领域已进入大数据时代, 然后由生物数据的组成特征及可视化的特点引出生物大数据可视化的重要性和必要性. 本文总结了生命科学研究领域中不同类型生物大数据的可视化研究进展, 最后讨论了目前生物大数据可视化所面临的挑战, 并提出可能的解决方案.
关键词 大数据生物信息学可视化
2005年以来, 随着高通量测序技术的不断进步及广泛应用, 生命科学跨入了大数据时代. 以基因组科学和生物医学领域为代表的生命科学研究产生和积累了海量的数据信息: 欧洲生物信息学研究所 (European Bioinformatics Institute, EBI)目前存储了将近20 PB的数据, 其中基因组数据约占2 PB, 这一数字随着新一代测序技术的不断发展每年成倍增长[1]; 高通量测序数据库(Sequence Read Archive, SRA)作为美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)最主要的高通量数据存储库, 目前存储的数据总量超过了3 PB, 对外发布的数据量达到1640 TB[2]; 此外, 当前世界上最大的基因数据产出机构——华大基因研究院(Beijing Genomics Institute, BGI)每天产出包括人、植物、动物和微生物在内的约6 TB基因组数据[1].
国际上的多个合作研究项目产生了史无前例规模的生物数据. 为了破译人类的全部遗传信息, 美国科学家在1985年率先提出了人类基因组计划(Human Genome Project, HGP)[3], 这一计划不仅覆盖了 99.99%的人类基因组, 解读了人体基因密码的“生命之书”, 而且推动了生命科学和生物技术的基础性研究, 促进了一系列科学技术的产生和发展; 2004年, 为了寻求新一代DNA研究技术对人类基因调控序列在全基因组水平上研究的应用, “DNA元件百科全书” 计划(Encyclopedia of DNA Elements, ENCODE)启动, 这一计划促使来自32个科研机构的442名研究人员获取并分析了超过15 TB的原始数据[4]; 从2005年底至今, 由美国国国家癌症研究所(National Cancer Institute, NCI)和美国国家人类基因组研究所(National Human Genome Research Institute, NHGRI)共同发起的癌症基因组图谱计划(The Cancer Genome Atlas, TCGA)[5], 通过多个基因组技术平台分析并获取超过 800 TB数据及文档资料, 为诊断、治疗和预防癌症打下了敦实的基础; 2010年, 中、英、德、美等国共同合作了国际千人基因组计划(1000 Genomes Project), 至今为止产生的数据量达到50 TB, 其中包含来自全球27个族群的2500个人的全部基因组信息[6].
当今测序技术的进步速度之快, 已远超计算机领域里的摩尔定律[7](价格不变时, 集成电路的性能每18个月增加一倍). 在1990年启动的人类基因组计划中, 美、欧、中、日等多个国家和地区超过200名科学家, 投入了超过10年的时间和约30亿美元才完成人类全基因组的测序; 但现在, 仅靠一个实验室的数名研究人员, 就可在数周内完成人类全基因组重测序, 而试剂成本则可控制在1000美元之内. 如此巨大的进步, 不仅给生命科学的研究带来了巨大的机遇, 在此基础上如何有效处理和分析这些测序数据, 也给此领域内的研究人员带来了巨大的挑战.
以DNA序列数据为代表的海量数据是构成生命科学研究的重要组成部分, 通过应用生物信息学技术进行大数据研究, 理解隐藏在大数据里的生物学知识成为当前生物技术发展的迫切需求. 传统的基于文本的数据处理和展示模式已经严重制约了对于生命科学大数据的解读. 基于可视化技术的信息挖掘成为一种必需的解决途径. 可视化是对事物建立心理模型或者心理图像的一个过程1). 通过可视化, 抽象的符号信息可以转化为易于理解的图像和模型, 另外交互式的使用允许研究人员从不同的可视化角度来探究隐藏在大数据里的不同模式和关联. 可视化拥有强大的将复杂数据转化为可利用信息的能力. 生物数据复杂、冗余等诸多特点决定了可视化是有效地理解生物数据的不可或缺的手段. 生物大数据可视化依托于现有的计算技术, 在一定时间内产生视觉表现模型, 并在此基础上尽可能地增强交互性, 从而加强用户体验以及对生物数据分析结果的认知能力.
1 生物大数据的特征及来源
生物大数据除了具有传统大数据4“V”的特点, 即数据量大(Volume)、数据处理速度快(Velocity)、数据源多变(Variety)和蕴含价值(Value)外[8], 还拥有其特有的数据复杂性(Complexity)[9]. 有生物学家提出, 复杂程度将生物领域产生的大规模数据与其他科学领域的产出区分开来. 在高能物理中, 数据有着合理的结构和注释, 而生物学数据目前来讲却难以完美地组织起来. 除了简单的基因组测序外, 生物学家会追踪许多不同的细胞和分子成分, 试图使用各种手段弄清其中包含的复杂关系. 此外, 由于生物数据经常来自不同的实验方法和机构, 使用了不尽相同的参数标准, 产生的数据类型丰富多样, 导致这些数据可能采用不同的存储结构(如narrowPeak, BED, SAM 等), 针对不同的研究对象(如基因序列、蛋白质互作关系、菌群共生等), 来源于不同的渠道(如测序、医疗记录等). 不同实验的参数标准、特异的细胞组织类型以及无法结构化存储的药物处理过程等诸多因素都是造成生物大数据复杂性的原因. 生物数据可视化的核心就是利用有效的算法消除这些数据的复杂性, 从而将其中隐含的生物学规律清晰地展示给用户, 而解析、转换这些复杂数据的格式, 则是数据可视化设计的第一步, 下面以数据来源为分类来了解生物数据复杂多样的格式特征.
首先, 测序技术的飞速发展为生物领域提供了数目庞大的宝贵资源. 目前第二代测序技术被广泛采用, 第二代测序产生数以百万计的短序列, 再由拼接算法将这些短序列在全基因组范围内组装起来, 从而进行进一步的数据分析工作[10]. 迄今为止, 新兴的单细胞测序技术一直被认为是最为值得关注的测序技术, 传统的测序方法忽略了细胞间的差异性, 得到的结果仅仅是一群细胞信号的平均值, 而基于单细胞水平对全基因组进行扩增与测序的单细胞测序技术, 不仅在基因表达量方面测量精准, 而且能够检测到表达量较低的基因及非编码RNA, 因此具有很大的优势及发展空间[11]. 除此之外, 单细胞 RNA测序(single-cell RNA-seq)使追踪单个细胞的转录组成为可能 [12], 染色质免疫共沉淀测序 (ChIP-seq)[13]等实验技术有力地支持了对基因组数据的功能性注释. 这些高通量的测序技术, 为研究者发现与疾病相关的基因型变异、研究某个表型的整个转录组、某一条件下的甲基化状态以及对DNA上蛋白质结合位点进行定位等工作提供了便利与支持, 然而随着数据规模的增大, 测序数据的处理和分析逐渐成为瓶颈.
其次, 生物芯片技术的使用在过去的数年中产生了庞大的数据资源. 为了实现对生物组织、细胞、蛋白质、核酸等组分中富含的大量信息进行快速准确的检测, 研究人员在固体芯片表面构建了微型的生物化学分析系统. 当前的生物芯片主要分为微阵列芯片和微流控芯片两种类型[14]. 传统的以静态和杂交技术为基础的微阵列芯片主要有基因芯片(DNA Microarray)、蛋白芯片(Protein Chip)和芯片实验室 (Lab-on-a-chip)等形式[15]. 其中, 基因芯片也叫DNA 高密度微点阵杂交技术, 以核酸探针互补杂交技术为基础而建立, 可用于DNA序列测序、基因表达分析、基因分型以及基因多态性分析等研究目的; 蛋白芯片依据蛋白质分子和其他分子的相互作用而构建; 而芯片实验室将整个流程集约化形成微型的分析系统. 芯片与生物分子反应所产生的信号需要借助于芯片扫描仪, 并通过相关软件分析采集到的各反应点的荧光强弱信号、所在位置信息所形成的图像来获取有关的生物信息. 微流控芯片以微流体控制技术为基础, 主要有毛细管电泳芯片、PCR反应芯片等形式[15]. 近年来, 生物芯片技术在基因表达水平检测、基因诊断、药物筛选、个体化医疗临床、疾病诊断和治疗、疾病易感基因发现以及基因功能确认等医学与生物学领域得到广泛的应用.
再次, 生物质谱为生命科学的研究做出了巨大的贡献, 不仅被认为是大规模、高通量鉴定几十万分子量的生物大分子结构的首选工具, 而且对于研究蛋白-蛋白等大分子之间的相互作用、翻译后修饰以及基因表达水平的变化有着很大的帮助. 质谱法主要原理是先将样品变为气态的离子混合物, 再按照质荷比(m/z)进行分离, 从而成功获取样品的质量、含量及结构等信息[16]. 在获取使用谱图法或列表法表示的测定结果后, 需要进行进一步的数据分析. 对于鉴定蛋白质的方法, 目前常用的有质量纹鉴定法 (Peptide Mass Fingerprinting)、二级质谱的数据库搜索鉴定法(MS/MS Database Searching)等手段[17]. 质谱分析技术被称作蛋白质组的核心技术, 最近在 Nature上公布的人类蛋白质组草图就是基于16857个质谱分析实验结果的整合[18]. 基质辅助激光解析-飞行时间质谱系统(VITEK®MS)作为美国FDA批准的首个用于检测病菌的质谱检测系统, 可用于酵母菌和致病细菌临床快速鉴定, 这也是第一种能在数分钟内检测致病微生物的医疗器械[19].
此外, 通过各种先进手段获取的与生物相关的图片影像资料也日益丰富起来. 生命体内存在着蛋白质、RNA以及DNA等种类繁多的生物大分子. 随着显微镜、成像捕捉等高精尖端仪器技术的不断发展, 科学家们不仅能够通过低温电子显微镜直接观察到蛋白质等生物大分子精细到原子的组织结构, 而且逐渐可以直接观测记录到活体组织中生物大分子在时间、空间维度上的结构变化和各分子间的相互作用的动态画面. 目前, 美国斯坦福大学研究人员借用 “微型内窥镜”及玻璃导管已经实现了在不破坏活体被观察组织的情况下, 长时间地对活体大脑神经元进行观测[20]; 北京大学开发的“生物正交受激拉曼散射成像”技术成功地特异性标记了活细胞的脂类、核糖、蛋白质和糖类等成分[21]; 美国纽约冷泉港实验室将分子标记手段与显微镜技术相结合, 顺利完成了第一个活体老鼠体内肿瘤细胞活动的影像记录工作[22]. 通过这些高新技术手段, 科学家们有望从中得到所有细胞、组织中蛋白质和复合物的相关位置, 弄清人体的有机物概况. 因此, 越来越多非结构化的图片影像数据亟待批量化整合、分析及展示.
最后, 临床数据也是一个不可忽略的数据来源. 仅隶属于中国中医科学院的广安门医院每年产生的数据量高达就70 TB2), 如果将全国的临床数据都集合在一起, 其数据规模更是不可估量. 现有的临床医学数据包含电子病历、医学影像、化验结果以及生化检查、病理切片检查的生物学信息等, 这些临床信息不但多样、冗余、不完整, 而且往往涉及患者隐私、公司利益冲突等问题, 加之有些数据之间难以关联, 造成标准化实施的困难. 这种结构化与非结构化格式并存的特点, 使得临床数据的整理分析变得异常困难[23]. 为了挖掘这些医疗数据中潜在的价值, 一些临床和科研机构着手将医疗数据进行整合, 构建临床试验数据的共享和分析平台. 北京的各大医院通过临床科研信息共享系统将实践数据化、规范化、数字化, 海量的数据通过整理转换等过程, 被进一步应用在查询检索、统计分析和数据挖掘上, 以此获取新的知识, 从而更加有效地对临床实践进行指导2). 美国临床肿瘤学会(American Society of ClinicalOncology, ASCO)旗下的“CancerLinQ”允许研究人员进入、访问和分析匿名癌症患者的病例[24]; 新型的电子诊断领域也为信息整合提供了极大的便利. 海量的临床数据的整合利用将大大有助于科研人员及医学专家对大规模疾病患者群体治疗情况进行分析, 从而为攻克疑难杂症提供契机.
相关知识推荐:收数据可视化论文的期刊
除了上述几个主要的生物大数据来源以外, 新型的技术手段不断贡献出宝贵的资源数据, 例如最新的流式荧光技术[25]可以实现快速、准确、高通量地对肿瘤标志物进行检测, 此外不同类型的仪表设备也为生物领域提供了不少有价值的数据. 丰富的数据来源显示出生物数据不仅数据规模庞大, 类型复杂多变, 而且在立体空间上结构、位置随时间不断变换、移动. 解决这些数据的存储只是最基本的任务, 更为重要的是使用这些数据. 同样, 对生物大数据进行可视化是为了更加充分地挖掘出数据中潜在的价值, 因此在设计可视化工具时如果能够以数据来源为依据, 从数据规模、复杂度、空间性和时间变换性这4个方面针对目标数据进行考虑, 将十分有益于从数据中获取有效信息.
2 生物大数据可视化类型及现状
可视化对生物数据的分析至关重要, 以生物数据的特性来看, 一般情况下仅凭文字很难描述清楚其中存在的复杂关系. 可视化不仅可以用来进行形象展示, 更是数据分析的第一个战场, 对生物数据进行良好的直观、交互性展示可以揭示出数据内在的错综复杂的关联状况, 在这一点上其他方法很难与可视化相提并论. 从最简单的Excel电子表格、Google 文档到R, Pandas等统计编程架构, 再到D3.js, Prefuse 等可视化程序包, 这些通用数据可视化和处理工具都可以为数据分析、信息挖掘提供很好的计算机手段. 另外针对于不同的数据类型和目的, 生物领域涌现了一大批开源、优秀的可视化工具(图1), 这些针对生物研究人员开发的工具易于上手, 为生物数据的快速分析提供了便利.
2.1 测序数据
测序技术、生物芯片提供了存在于生命体中的 DNA, RNA, 蛋白质等大分子的丰富的一级序列资源, 现有的基因组浏览器根据不同的需求对这些基序进行了从细节到宏观的展示. 以当前最为常用的 UCSC Genome Browser[26]为例, 它支持可以被比对到基因组上的任何数据类型, 将图像在服务器端渲染后嵌入网页中. 它对于基因组数据的展示模式体现了大多现存浏览器共有的特点: (1) 以染色体位置为索引的基因组数据视图; (2) 以参考基因组为标准提供位置坐标轴; (3) 基于track展示; (4) 良好的交互性和可定制性, 可根据用户所需进行装载或隐藏数据内容. 除了这些展示特征外, 不同的基因组浏览器也拥有自己独特的功能. GenomeView[27]提供注释编辑器, 可以展示和注释信息, 进行多序列比对、共线性匹配、短序列比对以及其他可以被显示的内容; 交互探究大型集成数据集的可视化工具 (Integrative Genomics Viewer, IGV)[28]可支持多种数据类型的交互展示, 包括测序序列比对、基因表达数据和拷贝数异常(图1(e))等.
由于不同的组织转录组的表达差异往往借助于统计手段进行聚类, 并需要使用热图使聚类结果呈现直观的展示, 并加以解释, 聚类得到的不同表达模式还可进一步按照功能富集程度进行分类并以图形化方式表示假设检验的结果, 以Gitools[29]为代表的此类工具采用了热图的形式对基因组数据进行集成化分析和展示, 此工具通过引入KEGG, Biomart等生物数据库达到对先验知识的利用, 提供富集分析、相关性分析以及显著性计算等丰富的分析手段, 通过集成排序、过滤、移动、聚集、搜索及可视化行列注释等功能允许使用者交互性地分析和可视化多维数据.
此外, 测序数据的可视化可能会对数据的深入挖掘起着决定性的作用. 例如, 单核苷酸多态性 (SNP)、插入缺失标记(InDel)以及基因组结构变异是一级序列中颇受关注的内容, 它们往往与复杂疾病的发生发展有着密切关系. 其中, 基因组结构变异包括插入、删除、倒置、易位、复制以及拷贝数变异等不同的类型, 每种类型使基因组产生不同的结构改变. 由于各类结构变异的复杂性, 以及真核生物基因组结构固有的重复序列特性, 导致仅凭现有的算法很难完全正确地检测出每种类型的变异. 特别地, 结构变异往往会引起短序列的错误定位, 进而导致小尺度的多态性预测错误, 因此通过提供可视化工具来方便研究者进行人工判断在结构变异的检测和识别中变得不可或缺[30]. 目前已有诸多的致力于展示、探究结构变异的可视化工具, 如可以运行在各种操作系统上的针对结构变异的集成软件inGAP-sv[30], 不仅能够以较低的假阳性概率检测出复杂的变异类型, 而且提供了友好的可视化接口, 每种类型结构变异特征模式进行标识, 通过右击鼠标可获取关于特定读长或结构变异的所有信息(图1(f)). 除此之外, inGAP-sv允许使用者根据自身的需求灵活设定显示测序短序列的外型和连线的颜色, 以便更好地为探究结构变异提供便利. inGAP-sv针对于结构变异提供识别、可视化、注释、人工编辑等一站式的服务, 这种集可视化、挖掘为一体, 注重用户体验度的工具设计方式预示了未来的软件开发走向.
2.2 分子结构数据
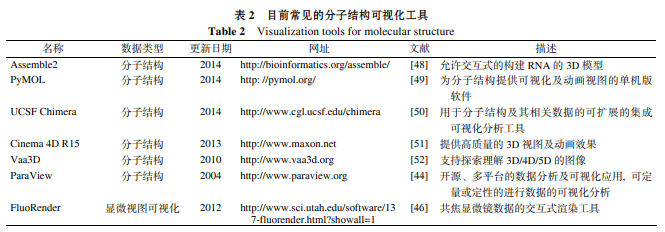
结构分子生物学是将物理和化学与生物学相连接的一门关键学科, 它主要聚焦于3D和4D复杂形状和功能关系的研究, 荧光标记、显微观察以及成像捕捉等技术为这一领域提供了丰富的视图数据, 而众多服务于分子结构的可视化工具在研究过程中起到了极大的作用. 以可视化软件ParaView[44]为例, 它允许使用者通过定性和定量的技术手段对大量的数据集快速建立3D视图模型, 从任意的角度对分子结构进行观察. 由于蛋白质等大分子结构精细复杂, 其内部的位置关系需要大量的计算资源, 因此3D视图软件往往比2D展示工具需要更加高效的算法设计, 高性能的计算设备以及高分辨率的展示屏幕. 为了增强对大规模数据集的处理能力, ParaView使用了分布式存储计算资源, 可以运行在超级计算机上来对万亿次级的数据集进行可视化分析. 除了ParaView, Amira[45], FluoRender[46]等工具都可以用来浏览分析 CT, MRI和显微图像, 以及实现对分子结构的3D 还原.
这些以计算图形学为基础所开发的软件工具虽然以更为精细准确的展示方式取代了物理模型, 但是却失去了与物理对象互动接触时产生的固有的视觉丰富性, 而这种触觉和本体感受往往为理解3D模型和进行物理操作提供了关键的线索. 因此工业领域的物体分层制造逐渐被应用在对分子结构的还原上. 譬如最近清华大学与美国德雷克赛尔大学研究人员以混合胶、纤维蛋白和宫颈癌细胞为原材料, 在精准的参数控制下, 利用一台3D细胞打印机成功制造出了与自然肿瘤十分接近的肿瘤模型[47].
2.3 关系网络
生物领域中由于生物分子互作、代谢途径、调控作用和基因表达等现象的存在促使了各种各样的关系网络的存在, 随着科学家们对这些过程的深入研究, 人们对其复杂度的了解也在不断增加. 生物学家经常需要对此类有相互作用关系的复杂系统和高维数据进行分析, 因此产生了可以对各种网络关系进行可视化的软件工具. 目前常用的复杂网络可视化工具有Cytoscape[53]、R中的igraph包以及Perl中的 GraphViz包等. Cytoscape代表一类以点线模式为基础进行网络可视化的工具, 它提供基础的功能布局和网络查询功能, 并且能够依据基本数据关系动态生成可视化网络. 其中因子、蛋白质和分子使用点表示, 两点间的交互关系用连接也就是边进行表示. 这种表示模式整合了分子间相互作用的网络, 适用任何分子系统的结构和相互关系, 允许将蛋白质、DNA 和其他对人类和生物有重要作用的分子数据库关联起来, 形成庞大的网络结构. 此外, R中的NetBioV, Gephi[54](图1(c))等软件包为生物信息学者提供了对节点连接类型的网络关系可视化开发工具.
随着计算手段的进一步发展, 网络关系的3D可视化形式逐渐发展起来. BioLayout Express3D[55]可以用于在2D, 3D空间内的可视化、分类归纳、探索和分析大型的网络关系. 此软件可对蛋白质互作和序列相似性等关系形成的网络进行展示, 摒弃了传统的对微阵列基因表达数据进行统计学差异分析的方法, 转而基于关联度评估来定义表达量间的相似性, 从而形成数据分析的网络范式 , 而且此工具基于 OpenCL并行框架编写, 充分考虑到网络关系3D可视化时所需的计算资源及图形处理技术支持等问题. 在2D或3D环境中BioLayout Express3D提供以下3个功能; (1) 对图像的移动、翻转和缩放操作; (2) 节点、边的个性化定制, 且允许设定文本标签以加强示意功能; (3) 背景颜色、3D灯光和投影、节点表面纹理等显示内容可进行偏好设置, 以便更好地对可视化效果进行渲染.
2.4 临床数据
虽然电子病历的使用范围在不断地扩大, 但是不统一的标准、非结构化的数据模式对研究者获取疾病治疗的真实资料造成了很大的障碍. 科学家们也开始着手处理这个问题, 以整合人类肿瘤数据为目标的Flatiron就是其中一个代表, Flatiron搭建的基于云端的OncologyCloud[63]平台聚合并转换了来自多渠道的患者信息、药单信息和患者恢复状况等数据, 并提供对数据集的归纳分析(图1(d)), 由此医生不仅能够通过OncologyCloud看到同类患者的治疗结果, 还能追踪到以往不同治疗方案所产生的临床结果. 这样一个提供全面的肿瘤数据收集、分析的系统也为肿瘤领域的基础研究提供了极大的便利; “癌症生命科学协会CEO圆桌会(the CEO Roundtable on Cancer)” 推出的PDS计划(Project Data Sphere)[24], 尝试打造一个癌症三期临床试验数据共享和分析平台, 数据集由赛诺菲、辉瑞以及阿斯利康等机构共同提供, 这些数据集在去除患者个人信息后进行了统一编号. 由于旧习惯及某些规章制度的影响, 大量医疗数据的整合和挖掘还需时间来逐步发展和规范. 但不可否认的是, 将治疗信息汇集在一起进行分析展示对攻克疾病有着不可忽视的作用.
除了以上阐述的可视化工具, 根据不同的需求还存在着很多其他的可视化形式 (表 1~4). 例 如 , Chimera[50](图1(b))将分子结构和包括密度图谱、超分子装配、序列比对、轨迹在内的相关数据集成起来, 产生高质量的动画效果; 由于不同质谱仪所产生的蛋白质谱初始数据格式不同, 而蛋白质组学质谱数据分析中统计学算法的实现过于复杂, 数据表示可视化、特征提取可视化及分类可视化对蛋白质质谱数据的分析十分重要; 除此之外, 还存在针对于SNP展示、表观遗传学所提供的核小体定位及组蛋白分析结果的可视化、微生物群落概况的可视化分析、海藻图解等诸多专项专能的可视化软件工具. 生物大数据可视化工具种类繁多, 为了更好地为挖掘有效信息做铺垫, 其开发趋势向具有统计分析功能的一站式集成工具靠拢. 此外, 未来的生物大数据可视化工具在交互性、美观性、实用性方面会做得越来越好.
3 展望及未来的挑战
生物数据有着自己的特点, 不仅数据规模庞大, 分布在不同的组织机构, 而且维度高, 数据不完整性和不确定性强. 利用各种技术手段获取数据本身不是目的, 将数据进行可视化也不是目的, 真正的目的是探究生命的本质, 发现未知的规律, 为人类的健康幸福服务, 因此挖掘隐藏在数据背后的涵义成为生物信息学家们一致的目标. 充分了解目前在分析生物数据的道路上存在的一些挑战及潜在的解决方案具有重要的意义.
首先, 现有的海量生物数据中存在着大量的冗余和噪音, 生产数据的组织机构可以对原始数据进行标准化处理和质控. 例如, 可将数据分门别类, 使用统一的数据存储标准、规格等. 合理的预处理手段可在一定程度上降低数据规模及复杂度, 节省存储空间及数据传输成本, 同时也会提高数据的易读性, 减少研究者对数据进行相同处理所需要的计算时间和资源等.
其次, 由于产出的数据往往分布在不同的研究机构, 如何实现海量数据的共享是研究人员们普遍面临的一大挑战. 现有的分布式注释系统(DAS)[69]提供了一个潜在的解决方案. 它定义了一份用来交换基因或蛋白质序列及其注释的通信协议, 在此协议下, 基于网络的可视化系统可实现同一界面下对远程异地分布注释数据的可视化.
再者, 生物数据特有的复杂多样性给数据挖掘带来很大困难, 因此在对大批量数据进行可视化前, 数据投影及各种降低维度的技术被广泛采用. 与此同时, 人类视觉的敏锐性、使用者面对展示界面时的推断能力和信息搜索能力等因素都需要加以考虑. 对生物大数据进行可视化时, 需要记住目标使用者是人, 目的是信息的展示和探索, 而非一味地追求视觉美观. 在开发生物大数据的可视化工具时, 需要尽可能提高软件或平台的易用性, 充分考虑用户的体验度, 提供友好的交互界面.
此外, 在有限的时间内对大规模数据进行处理及可视化是最基本的要求. 除了通过使用优化算法对数据规模和可视化效率进行平衡外, 还可以引入并行处理技术. 在对若干数据集进行可视化时, 可将查询处理分散在多个并行节点上, 以此缩短运行时间, 加快可视化的速度.
除了前述内容, 用于传输生物数据的网络基础设施的建设、数据的存储方式等诸多方面都存在着一定的困难. 虽然在分析生物大数据的道路上面临着诸多挑战, 但是这些暂时的困难并不能阻止科学家们前进的脚步, 生命科学的神秘面纱最终将会在一代代科研人员的努力下被完全揭开.——论文作者:周琳① , 孔雷② , 赵方庆①*
* 稍后学术顾问联系您




