
X 射线光电子能谱仪的数据采集及处理技巧
分类:科技论文
时间:2021-09-10
摘要X射线光电子能谱(XPS)广泛应用于材料表面分析表征。在XPS分析过程中,对于经验不足的科研或者测试人员可能会引入一些主观性的判断,使得谱图测试及分析对数据分析质量产生影响。为获得更好的数据质量,本文根据国际标准和实际测试经验总结出一种通用于不同类型材料表面分析表征的XPS数据采集和处理分析思路,以方便测试人员能够更加系统的进行数据采集和分析,为材料研发人员提供更加全面的元素组成信息和谱图解析注意事项,从而更有效地助力材料表面分析研究,该方法对科研工作者分析准确度的提高具有重要的理论意义和实际应用价值。
关键词XPS数据采集数据处理
X射线光电子能谱仪(XPS)是材料表面分析中最常用的测试手段之一,由于XPS主要应用于开发、评估尚未成熟的新功能材料或者器件,科研人员往往需要通过XPS等测试手段对未知或者尚未足够了解的研发产品进行进一步了解,为材料的提升性能研究提供指导和方向。
随着高新技术的研发和注入,仪器测试也越来越智能化、自动化,测试人员可根据科研工作者的测试需求(只针对科研人员关注的元素)进行数据采集;而科研工作者也可以在对自己样品表面组成不是足够了解的情况下,直接对重点关注的元素数据进行分析。这为测试人员和科研人员带来简单快速便利的同时,也有可能会导致科研人员忽略仪器状态或者其他谱峰的干扰,为后期数据分析带来错误分析的隐患。测试人员如何发挥自己的岗位和专业优势快速有效的助力科研,为科研人员提供更加真实全面的样品信息以及更专业的数据解析指导,为材料研究和性能提升提供正确的指导方向,是值得仔细分析探讨的。
1测试前准备
仪器状态确认与定期维护良好的仪器状态是获得高质量数据的前提和保证,分析者并非一定需要在最佳性能(或者仪器极限)下进行数据测试,但他们可能需要好的重复性以及可接受的信噪比,因此对于测试人员来讲,有必要对仪器状态进行定期维护。常规仪器维护主要包括结合能标的校准、强度标重复性和线性、荷电校正等。
由于结合能是元素的指纹信息,XPS数据分析工作者可根据结合能进行元素或者化学态的定性判定。结合能标不准确或者误差太大会对数据分析带来很大干扰,甚至有可能会对材料研发工艺提供错误的指导方向。校准时需要使用纯的金、银、铜参考样品,GB/T22571-2017[1]对具体校准操作进行了介绍。需要注意的是在测试过程中需要定期记录CuL3VV俄歇峰或者Ag3d5/2、Au4f7/2和Cu2p3/2峰的结合能(GB/T22571-2017[1]标准建议每4个月进行一次),如有超出校准值,则需按照标准进行仪器结合能标的校准。
谱峰强度是用来测定样品表面元素组成的一个重要参数,主要包括强度标的重复性、强度标的强度值、强度标的线性。强度测量的重复性是仪器稳定性和一致性的一个重要体现;此外谱峰强度值需要在合适的强度区间,强度值过于微弱可能会导致重点关注的超低含量元素测量误差较大甚至对元素的存在与否做出错误判断,严重影响到材料表面的定性及定量研究,强度值过强可能会导致仪器强度标的非线性,从而影响到定量测量的准确性。其中GB/T28633-2012[2]和GB/T21006-2007[3]分别对强度标的可重复性和线性的测定进行了描述;此外随着测试时间的增加,仪器信号强度会呈现缓慢减弱的趋势,因此定期进行靶点更换是十分必要的(建议一年至少更换靶点一次)。
非导电样品或者导电效果不是十分良好的样品在X射线照射下会产生荷电,导致实际测试得到谱峰的峰位和峰形与非荷电状态下的测量结果产生较大偏差,严重时会影响到化学态的定性判定以及后期的定量分析。因此常规测试时一般会在开启中和枪的情况下进行,并且GB/T25185-2010[4]对荷电控制和荷电校正方法进行了详细描述。
2样品状态确认及样品制备
在获得样品时应与样品制备人员进行必要的讨论交流,以获得尽可能多的样品信息(如样品是否新鲜制备、样品形态、材料类型,测试目的以及重点关注信息等),这样测试人员会对样品是否会发生放气、降解以及样品磁性等做出判断,根据样品具体情况并结合GB/T28894-2012[5]标准进行样品前处理(如消磁、干燥、表面污染去除等),并根据客户测试需求进行测试项目的建议(如常规XPS点分析、变角XPS、成像、线扫描等)。
样品制备需要结合标准GB/T30815-2014[6],且制备过程中需要重点关注以下几点:(1)磁性样品应确保样品已经经过消磁处理;(2)需要将可能会发生放气、有磁性、易挥发以及含有容易造成交叉污染元素的样品进行单独制备;(3)对于多孔材料或泡沫金属样品建议压片制样;(4)样品尽量保证新鲜制备且污染程度较小,颗粒粒度较大时建议采用研钵进行研磨。
3数据采集
样品制备好被放置在仪器制备室并且真空达到要求后,可转移至分析室进行数据采集。通常情况下我们需要采集宽扫描谱(全谱),并根据宽扫谱图了解样品表面元素组成及大致含量,为后面高分辨窄扫谱图的数据采集参数做准备。
数据采集过程中需要注意以下几点:(1)消磁后的磁性样品数据采集应选择非磁性模式进行,以免对样品再次充磁;(2)对于含量较低且重点关注的元素一般可通过适当增加采集遍数的方式提高信噪比;(3)针对重点关注材料表面的定性分析研究,对分辨率要求不是太高的样品可通过增加通能方式,提高数据采集的灵敏度;(4)针对有机样品,在确保仪器真空度无明显变差(样品挥发导致)的前提下,尽快进行数据采集,尽量避免X射线辐射对样品造成的损伤;(5)对X射线敏感的样品(如光电材料),为保证表面荷电稳定,建议数据采集前用X射线辐照一段时间(如4min);(6)数据采集过程中,元素间存在谱峰重叠的可以通过加大数据采集范围将重叠谱峰一起采全,方便后面数据分析。
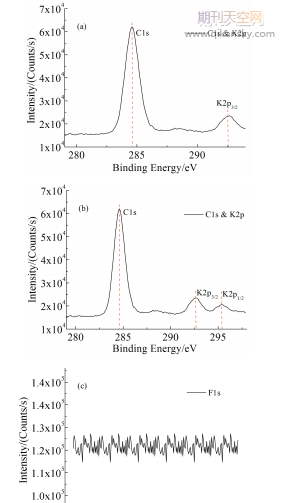
如图1(a)所示,由于在采集C1s高分辨谱图时发现在约292.5eV有谱峰存在,该处有可能为C-F化学态的C元素对应谱峰,也可能是来源于K元素的2p轨道对应谱峰贡献;然而由于C1s谱峰测试采集区域未能将K2p谱峰完全包含在内,这会影响到两元素的定性与定量分析。通过增加谱图采集范围将C1s和K2p谱峰采集完整(如图1(b)所示),可明显观察到K2p的双峰结构,且双峰面积比值约为2∶1,因此在293eV附近的谱峰并非出于C-F化学态下的C元素,而是K2p3/2的谱峰(此外由图1(c)可以进一步确认样品中确实不存在F元素)。
当同一区域内元素间的谱峰强度差异很大时(如图2(a)所示),由于O1s谱峰的存在导致508~528eV区域的V元素信号十分不明显,此时很容易忽略较低强度V2p谱峰的存在,从而影响该元素的定性判定以及与定量分析。当对508~528eV区域进行单独采集时,可明显观察到V元素对应的谱峰(如图2(b)所示),更方便对数据进行处理和分析。
(1)当某些元素的谱峰重叠严重,并且需要确认某种元素存在与否时,可采集该元素其他轨道的谱峰。以含P样品中Sr元素存在与否的判定为示例(如图3(a)所示),由于Sr3d与P2p产生严重的谱峰重叠,很难直接通过Sr3d与P2p数据对元素的存在与否做出定性判断,无法进行更进一步的定量分析。为对数据进行进一步确认,可对Sr3p轨道谱峰进行采集,如图3(b)所示,由于P2s谱峰区域为基线,则说明没有P元素的存在,因此在120~142eV区域全部为Sr元素的贡献。
如图4(a)所示,当Sr3p谱峰有较为明显的信号时,说明样品中确实含有Sr元素,因此图4(b)中在120~142eV区域中的谱峰信号来源于Sr和P两种元素共同的贡献,需要通过分峰拟合将各自谱峰进行拆分处理。
(2)有时候需要对特定元素进行化学态辨认,但元素XPS主峰结构峰位、峰形均对化学态不是十分敏感,此时需要采集部分特征峰进行指纹辨认。以Cu俄歇谱峰为例,如图5(a)所示,由于金属态和Cu2O化学态下Cu2p谱峰的峰位峰形极其相似,实际样品中无法根据Cu2p谱峰对Cu元素金属态和Cu2O化学态进行定性判定,此时结合图5(a)的Cu2p谱峰结合能峰位和5(b)的最尖锐俄歇谱峰的动能峰位计算得到的俄歇参数值可以为我们提供更加直接的化学态判定信息。
此外由于各化学态下的Mn2p谱峰的峰位、峰形极其相似,无法根据Mn2p数据对化学态进行判定,利用Mn3s的双峰劈裂间隔差值可以为Mn化学态的准确判定提供更加充分的证据(如图6所示)。
4数据处理(定性以及简单的定量分析)
4.1选择合适的背底扣除范围
通常情况下,材料制备研究人员会根据样品实际情况进行数据处理,因此在没有元素间的谱峰重叠的前提下,测试人员只需保证数据进行荷电校准和背底扣除、提供简单的定量结果即可。如图7所示,虽然样品中同时含有O元素和V元素,但O1s谱峰与V2p谱峰几乎无重叠,此时只需要将各自对应的谱峰进行定量即可(具体各元素的化学态情况可由科研人员根据自己实际材料情况进行确定)。
相关论文推荐:加强医用X射线摄影机质量控制
当存在谱峰重叠且不需要分峰拟合即可求得元素含量时,选择较为合适的背底扣除区域是十分必要的。由于样品中含有Na元素(如图8(a)所示),因此在493~499eV范围内Na的俄歇谱峰会与Sn3d3/2谱峰产生较为严重的谱峰重叠;此时并非一定需要通过分峰拟合方式扣除Na俄歇谱峰的干扰,可直接对Sn3d5/2谱峰进行背底扣除从而轻松快速获得Sn元素的相对定量结果。
4.2考虑利用其他轨道谱峰进行定量分析
如图9(a)所示,由于样品中含有大量的F元素,导致700~740eV区域出现较为明显的F的损失峰(谱峰较宽,且峰形不规则,无法通过常规数据处理方法进行分析);该谱峰的存在会对Fe2p谱峰的数据分析产生严重影响(如图9(b)所示),很难将F损失峰的贡献扣除,以获得较为精确的Fe元素的相对定量结果。此时可优先考虑采用常规拟合方法对Fe元素其他轨道如Fe3p的数据进行相对定量分析(如图9(c)所示)。
此外由于Fe3s较少受其他峰的干扰,且Fe3s的多重分裂也有助于对Fe元素的化学态进行判断,因此建议结合Fe3s谱峰进行定量以及辅助化学态判断。
4.3必要时的分峰拟合注意事项
当科研工作者需要通过分峰拟合方式获得更加真实的样品表面信息时,测试人员有必要提醒分析注意事项,如数据采集区域出现的谱峰并非全部来源于待测元素的贡献,有时其他元素会产生部分干扰,甚至有可能全部来源于其他元素的贡献。因此需要结合样品实际情况、元素以及元素其他轨道谱峰情况进行定性确认,这会对后续数据处理结果的准确性产生重要影响。
4.3.1确认无其他元素干扰的情况
如图10所示,该样品重点关注C元素所处的化学态以及对应的相对含量,通常情况下我们选择C1s作为C元素的主峰进行定性定量判定。但K2p3/2和处于C-F化学态下的C1s谱峰在291~296eV区域均会出峰,通过加宽C1s的谱图采集范围并同时采集F1s谱峰数据,可观察到并没有出现K2p的双峰结构,并且样品中含有明显的F元素。因此可以确定在数据显示此区域291~296eV区域为C-F化学态下的C1s谱峰(并不存在K元素的干扰),此时可直接通过背底扣除进行C元素的相对定量分析以及化学态确认研究。
4.3.2确认有其他元素干扰的情况
当样品重点关注Mo元素所处的化学态及对应的相对含量时,通常情况下我们选择Mo3d作为Mo元素的主峰进行定性定量判定。但由于S元素的存在(如图11(a)所示),导致在222~230eV区域会有S2s谱峰,此时进行Mo3d化学态的分析时需要将S元素产生的贡献进行扣除(如图11(b)所示)。
4.3.3确认无待测元素,完全为其他元素的贡献
样品表面为纯的Ni(OH)2(如图12(a)所示),由于Ni元素的存在导致770~815eV的范围内观察到十分明显的Ni俄歇谱峰(而此区域也恰好为Co2p谱峰的出峰范围,如图12(b)所示)。此时需要注意该区域信号主要来源于Ni元素的贡献,因此不可对Co2p谱峰采集区域进行背底扣除及Co元素的相对定量求取。——论文作者:范燕徐昕荣*石志锋李冰刘佳魏强鲁志龙孙成军
* 稍后学术顾问联系您




